Endonucleolytic cleavage site prediction using 5’-end RNA-Seq data
Complete source of this page is available in the notebook Endoribonucleolytic_sites_prediction.Rmd.
Based on the publication from Broglia L, Lécrivain A-L, Renault TT, Hahnke K, Ahmed-Begrich R, Le Rhun A, Charpentier E (2020) An RNA-seq based comparative approach reveals the transcriptome-wide interplay between 3′-to-5′ exoRNases and RNase Y. Nature Communications, 11, 1587. https://doi.org/10.1038/s41467-020-15387-6
Download datasets
Data from this article are publicly available on the SRA NCBI repository: https://www.ncbi.nlm.nih.gov/sra?term=SRP149896
For this notebook, we will use only 9 RNA-Seq samples corresponding to 9 datasets of Streptococcus pyogenes 5’-end RNA-Seq (SRX4173017, SRX4173016, SRX4173015, SRX4173014, SRX4173013, SRX4173012, SRX4173011, SRX4173010, SRX4173009).
As the datasets are too bug for direct download, fasterq-dump (instead of basic “curl/wget” command) from ncbi/sra-tools must be used to retrieve FASTQ files directly in the extdata directory of the GitLab project in the sub-directory called Spyo_Broglia where the reference genome is already provided when the project is clone:
for i in SRR7269351 SRR7269352 SRR7269353 SRR7269354 SRR7269355 SRR7269356 SRR7269357 SRR7269358 SRR7269359 ; do
fasterq-dump $i --outdir ../extdata/Spyo_Broglia ;
done
We remove all reverse files (rm SRR7236935._2.fastq.gz) and we gzip all forward files.
library(tidyverse)
time_start <- now()
library(rnaends)
Pre-processing
Thse datasets were already pre-processed when they have been submitted, so we can directly map the FASTQ files on the reference genome.
Quantification
We use the map_fastq_files to map the 9 FASTQ files in a single command.
bf <- map_fastq_files(list_fastq_files = list.files("../extdata/Spyo_Broglia/",
pattern = ".fastq.gz",
full.names = TRUE),
index_name = "../extdata/Spyo_Broglia/data/spyo_SF370.bt", # Path of the genome index (created if not exists)
fasta_name = "../extdata/Spyo_Broglia/spyo_SF370.fasta", # Path of the reference genome file
bam_dir = "../extdata/Spyo_Broglia/mapping_results", # Output directory
max_map_mismatch = 1,
aligner = "bowtie", # Either bowtie or subread
map_unique = FALSE,
force = TRUE,
threads = 3)
#> Warning in FUN(X[[i]], ...): Index directory already exists remove it if you
#> want to recreate it
#> Warning in FUN(X[[i]], ...): Index directory already exists remove it if you
#> want to recreate it
#> Warning in FUN(X[[i]], ...): Index directory already exists remove it if you
#> want to recreate it
#> Warning in FUN(X[[i]], ...): Index directory already exists remove it if you
#> want to recreate it
#> Warning in FUN(X[[i]], ...): Index directory already exists remove it if you
#> want to recreate it
#> Warning in FUN(X[[i]], ...): Index directory already exists remove it if you
#> want to recreate it
#> Warning in FUN(X[[i]], ...): Index directory already exists remove it if you
#> want to recreate it
#> Warning in FUN(X[[i]], ...): Index directory already exists remove it if you
#> want to recreate it
#> Warning in FUN(X[[i]], ...): Index directory already exists remove it if you
#> want to recreate it
bf
#> [[1]]
#> [1] "../extdata/Spyo_Broglia/mapping_results/SRR7269351_1.fastq.gz.bam"
#>
#> [[2]]
#> [1] "../extdata/Spyo_Broglia/mapping_results/SRR7269352_1.fastq.gz.bam"
#>
#> [[3]]
#> [1] "../extdata/Spyo_Broglia/mapping_results/SRR7269353_1.fastq.gz.bam"
#>
#> [[4]]
#> [1] "../extdata/Spyo_Broglia/mapping_results/SRR7269354_1.fastq.gz.bam"
#>
#> [[5]]
#> [1] "../extdata/Spyo_Broglia/mapping_results/SRR7269355_1.fastq.gz.bam"
#>
#> [[6]]
#> [1] "../extdata/Spyo_Broglia/mapping_results/SRR7269356_1.fastq.gz.bam"
#>
#> [[7]]
#> [1] "../extdata/Spyo_Broglia/mapping_results/SRR7269357_1.fastq.gz.bam"
#>
#> [[8]]
#> [1] "../extdata/Spyo_Broglia/mapping_results/SRR7269358_1.fastq.gz.bam"
#>
#> [[9]]
#> [1] "../extdata/Spyo_Broglia/mapping_results/SRR7269359_1.fastq.gz.bam"
Mapping statistics
The map_stats_bam_files function provides some basic statistics on the mapping:
map_stats_bam_files(bam_files = bf) %>%
knitr::kable()
#> BAM file mapping stats exists '../extdata/Spyo_Broglia/mapping_results/SRR7269351_1.fastq.gz.bam.map_stats.csv' already exists, set force=TRUE to recompute
#> BAM file mapping stats exists '../extdata/Spyo_Broglia/mapping_results/SRR7269352_1.fastq.gz.bam.map_stats.csv' already exists, set force=TRUE to recompute
#> BAM file mapping stats exists '../extdata/Spyo_Broglia/mapping_results/SRR7269353_1.fastq.gz.bam.map_stats.csv' already exists, set force=TRUE to recompute
#> BAM file mapping stats exists '../extdata/Spyo_Broglia/mapping_results/SRR7269354_1.fastq.gz.bam.map_stats.csv' already exists, set force=TRUE to recompute
#> BAM file mapping stats exists '../extdata/Spyo_Broglia/mapping_results/SRR7269355_1.fastq.gz.bam.map_stats.csv' already exists, set force=TRUE to recompute
#> BAM file mapping stats exists '../extdata/Spyo_Broglia/mapping_results/SRR7269356_1.fastq.gz.bam.map_stats.csv' already exists, set force=TRUE to recompute
#> BAM file mapping stats exists '../extdata/Spyo_Broglia/mapping_results/SRR7269357_1.fastq.gz.bam.map_stats.csv' already exists, set force=TRUE to recompute
#> BAM file mapping stats exists '../extdata/Spyo_Broglia/mapping_results/SRR7269358_1.fastq.gz.bam.map_stats.csv' already exists, set force=TRUE to recompute
#> BAM file mapping stats exists '../extdata/Spyo_Broglia/mapping_results/SRR7269359_1.fastq.gz.bam.map_stats.csv' already exists, set force=TRUE to recompute
bam_file |
mapped |
unmapped |
pc_mapped |
|---|---|---|---|
SRR7269351_1.fastq.gz.bam |
41814799 |
988534 |
97.69 |
SRR7269352_1.fastq.gz.bam |
41739505 |
938889 |
97.80 |
SRR7269353_1.fastq.gz.bam |
43357081 |
1019019 |
97.70 |
SRR7269354_1.fastq.gz.bam |
43026077 |
1016127 |
97.69 |
SRR7269355_1.fastq.gz.bam |
34567930 |
650684 |
98.15 |
SRR7269356_1.fastq.gz.bam |
37802063 |
705215 |
98.17 |
SRR7269357_1.fastq.gz.bam |
26960617 |
641089 |
97.68 |
SRR7269358_1.fastq.gz.bam |
34908063 |
977130 |
97.28 |
SRR7269359_1.fastq.gz.bam |
40858741 |
759768 |
98.17 |
Quantification (counts computation)
In order to obtain counts per position, we use the quantify_from_bam_files function to parse previoulsy obtained bam_files.
This function allows the quantification of either the 5’-ends (quantify_5prime) or the 3’-ends (quantify_3prime), in all cases it’s an exact position quantification.
Run with minimal parameters:
count_table <- quantify_from_bam_files(bam_files = bf,
sample_ids = c("WT1", "WT2", "WT3",
"Y1", "Y2", "Y3",
"suppl1", "suppl2", "suppl3"), # Set an identifier to know the source of each values
fun = quantify_5prime,
force = TRUE)
count_table %>%
head(15) %>%
knitr::kable()
sample_id |
rname |
position |
strand |
count |
|---|---|---|---|---|
WT1 |
AE004092.2 |
38 |
+ |
2 |
WT1 |
AE004092.2 |
205 |
+ |
819 |
WT1 |
AE004092.2 |
206 |
+ |
13 |
WT1 |
AE004092.2 |
211 |
+ |
7 |
WT1 |
AE004092.2 |
215 |
+ |
8 |
WT1 |
AE004092.2 |
217 |
+ |
34 |
WT1 |
AE004092.2 |
218 |
+ |
2 |
WT1 |
AE004092.2 |
219 |
+ |
2 |
WT1 |
AE004092.2 |
219 |
- |
5 |
WT1 |
AE004092.2 |
236 |
+ |
15 |
WT1 |
AE004092.2 |
248 |
+ |
31 |
WT1 |
AE004092.2 |
253 |
+ |
13 |
WT1 |
AE004092.2 |
254 |
+ |
1 |
WT1 |
AE004092.2 |
262 |
+ |
14 |
WT1 |
AE004092.2 |
265 |
+ |
117 |
Downstream analysis
Differential abundances for prediction of cleavage sites
As in the article, we’re going to carry out a differential gene expression analysis (DGE), but at the resolution of an exact position to detect RNase Y endonucleolytic cleavage sites by comparing 3 S. pyogenes strains:
Wild-type
RNase Y deletion mutant
RNAse Y deletion mutant supplemented
To perform a DGE analysis the “edgeR” package is be used (which is not part of the requirements of the rnaends package).
We use as input data the count table we’ve just produced.
library(edgeR)
#> Loading required package: limma
Pivot the count table to create a DGEList object
group1 <- c("WT", "WT", "WT")
group2 <- c("Y", "Y", "Y")
group3 <- c("suppl", "suppl", "suppl")
# Pivot count table
data <- count_table %>%
pivot_wider(id_cols = c(position, strand),
names_from = sample_id,
values_from = count, values_fill = 0)
# Transform into matrix for DGElist
mat <- data[,3:ncol(data)] %>%
as.matrix()
rownames(mat) <- paste0(data$position, data$strand)
mat %>%
head(15) %>%
knitr::kable()
WT1 |
WT2 |
WT3 |
Y1 |
Y2 |
Y3 |
suppl1 |
suppl2 |
suppl3 |
|
|---|---|---|---|---|---|---|---|---|---|
38+ |
2 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
205+ |
819 |
491 |
358 |
1001 |
345 |
568 |
212 |
304 |
976 |
206+ |
13 |
22 |
5 |
44 |
17 |
117 |
3 |
4 |
21 |
211+ |
7 |
11 |
5 |
35 |
0 |
0 |
1 |
1 |
0 |
215+ |
8 |
1 |
10 |
0 |
4 |
4 |
1 |
0 |
0 |
217+ |
34 |
13 |
7 |
16 |
2 |
8 |
15 |
1 |
10 |
218+ |
2 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
219+ |
2 |
0 |
1 |
0 |
0 |
0 |
0 |
0 |
0 |
219- |
5 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
236+ |
15 |
0 |
0 |
1 |
0 |
1 |
0 |
0 |
31 |
248+ |
31 |
15 |
2 |
6 |
0 |
9 |
45 |
0 |
1 |
253+ |
13 |
0 |
0 |
0 |
0 |
1 |
0 |
0 |
0 |
254+ |
1 |
0 |
0 |
3 |
0 |
0 |
0 |
0 |
0 |
262+ |
14 |
0 |
6 |
1 |
3 |
14 |
43 |
0 |
0 |
265+ |
117 |
33 |
1 |
0 |
15 |
11 |
0 |
0 |
0 |
DGEList and filter count according cpm
dge <- DGEList(counts = mat[,1:9], group = factor(c(group1, group2, group3)) )
# Keep position where there is at least 6 conditions with cpm>0.05
keep <- rowSums(cpm(dge) >= 0.05) >= 6
dge.filter <- dge[keep, ]
# Update dge (for racalcul of cpm)
dge2.filter <- DGEList(dge.filter$counts,
group = factor(c(group1, group2, group3)) )
# Keep position where there is at least 3 conditions with cpm>5
keep <- rowSums(cpm(dge2.filter) >= 5) >= 3
dge3.filter <- dge2.filter[keep, ]
cpm(dge3.filter) %>%
head(15) %>% knitr::kable()
WT1 |
WT2 |
WT3 |
Y1 |
Y2 |
Y3 |
suppl1 |
suppl2 |
suppl3 |
|
|---|---|---|---|---|---|---|---|---|---|
205+ |
22.170898 |
13.4253747 |
9.1030977 |
27.186211 |
10.908131 |
16.3304562 |
8.5810845 |
9.3700179 |
26.9990941 |
2540+ |
5.820199 |
2.1054050 |
7.6028665 |
1.249316 |
1.359564 |
2.5588215 |
9.9572962 |
2.3425045 |
0.2489671 |
3452+ |
132.483974 |
189.8965934 |
68.4003711 |
212.057875 |
77.115743 |
78.1446831 |
47.9650241 |
67.0079572 |
177.7071523 |
4499+ |
6.253330 |
8.1481908 |
2.5173371 |
8.609419 |
1.960302 |
1.3225369 |
3.0357610 |
1.1712522 |
0.0276630 |
4512+ |
20.790293 |
7.3825889 |
7.2977348 |
14.828842 |
13.121375 |
4.6576301 |
11.6573223 |
7.3665602 |
0.1659780 |
11902+ |
12.777367 |
6.4255867 |
12.4341195 |
28.082459 |
14.259615 |
15.2954273 |
2.0238407 |
6.8117565 |
26.1138779 |
12203+ |
10.124439 |
8.6403634 |
4.2718447 |
4.725675 |
6.797821 |
6.6414355 |
1.4571653 |
7.6131396 |
0.8852162 |
13365+ |
8.418986 |
3.1991219 |
4.3481277 |
5.377492 |
7.493412 |
3.7376044 |
3.3190987 |
0.7089158 |
4.5367330 |
13441+ |
5.874340 |
6.9997880 |
3.6107259 |
5.784878 |
2.719128 |
3.3350932 |
4.4119727 |
0.2157570 |
0.0000000 |
13446+ |
12.750297 |
5.7693565 |
7.7554324 |
9.695781 |
4.584577 |
8.2514805 |
24.4075186 |
1.5719438 |
0.0276630 |
13451+ |
8.121208 |
0.1093717 |
9.4590848 |
7.197149 |
1.201475 |
0.5750161 |
3.4405292 |
0.3698691 |
0.2766301 |
13475+ |
12.235954 |
24.9094020 |
3.1530283 |
8.745215 |
4.963990 |
5.3764002 |
4.4119727 |
3.6062240 |
23.5412183 |
13527+ |
1.840807 |
0.2460863 |
0.9916782 |
14.204184 |
5.469874 |
6.6126847 |
0.0404768 |
0.3082243 |
21.0792108 |
13552+ |
17.541809 |
6.7263588 |
0.9662506 |
8.364988 |
7.019145 |
5.4626526 |
8.0548859 |
0.4931588 |
0.0276630 |
13559+ |
6.172118 |
3.8553520 |
0.3559871 |
16.376908 |
1.517653 |
4.1113649 |
4.2905422 |
0.0616449 |
9.3224331 |
Normalize and estimate dispersion
Normalization
dge3.filter <- calcNormFactors(dge3.filter, method = "TMM")
Dispersion
disp <- estimateDisp(dge3.filter)
#> Using classic mode.
Exact tests
Comparison WT vs RNase Y deletion mutant
et <- exactTest(disp, pair = c("WT", "Y"))
res_WT <- et$table
res_WT$fdr <- p.adjust(res_WT$PValue, method = "fdr") # Adjusting p.values
res_WT$Position <- as.integer(str_sub(rownames(res_WT), 1, -2)) # Retrieving positions using stocked into the rownames
res_WT$strand <- str_sub(rownames(res_WT), -1) # Retrieving strand
res_WT %>%
head(15) %>%
knitr::kable()
logFC |
logCPM |
PValue |
fdr |
Position |
strand |
|
|---|---|---|---|---|---|---|
205+ |
0.4757538 |
4.097125 |
0.6161737 |
1.0000000 |
205 |
+ |
2540+ |
-1.2969346 |
1.768362 |
0.3370351 |
1.0000000 |
2540 |
+ |
3452+ |
0.0168134 |
6.936504 |
0.9846036 |
1.0000000 |
3452 |
+ |
4499+ |
-0.4542850 |
1.736353 |
0.7499819 |
1.0000000 |
4499 |
+ |
4512+ |
0.0787080 |
3.143991 |
0.9482911 |
1.0000000 |
4512 |
+ |
11902+ |
1.0497216 |
3.917431 |
0.3388455 |
1.0000000 |
11902 |
+ |
12203+ |
-0.0990961 |
2.468814 |
0.9318575 |
1.0000000 |
12203 |
+ |
13365+ |
0.2956658 |
2.204602 |
0.8001578 |
1.0000000 |
13365 |
+ |
13441+ |
-0.3248103 |
1.752619 |
0.8400716 |
1.0000000 |
13441 |
+ |
13446+ |
0.0078938 |
2.913511 |
0.9962916 |
1.0000000 |
13446 |
+ |
13451+ |
-0.8812748 |
1.583629 |
0.5850758 |
1.0000000 |
13451 |
+ |
13475+ |
-0.9495031 |
3.505009 |
0.4234994 |
1.0000000 |
13475 |
+ |
13527+ |
3.3057756 |
2.826576 |
0.0722374 |
0.5765283 |
13527 |
+ |
13552+ |
-0.0277194 |
2.451620 |
0.9866344 |
1.0000000 |
13552 |
+ |
13559+ |
1.1973548 |
2.436253 |
0.4545305 |
1.0000000 |
13559 |
+ |
Comparison supplemented RNase Y deletion mutant vs. RNase Y deletion mutant
et <- exactTest(disp, pair = c("suppl", "Y"))
res_suppl <- et$table
res_suppl$fdr <- p.adjust(res_suppl$PValue, method = "fdr") # Adjusting p.values
res_suppl$Position <- as.integer(str_sub(rownames(res_suppl),1,-2)) # Retrieving positions using stocked into the rownames
res_suppl$strand <- str_sub(rownames(res_suppl),-1) # Retrieving strand
res_suppl %>%
head(15) %>%
knitr::kable()
logFC |
logCPM |
PValue |
fdr |
Position |
strand |
|
|---|---|---|---|---|---|---|
205+ |
-0.2700981 |
4.097125 |
0.7756757 |
0.9841472 |
205 |
+ |
2540+ |
-1.1317669 |
1.768362 |
0.4033664 |
0.8990873 |
2540 |
+ |
3452+ |
-0.2658754 |
6.936504 |
0.7586034 |
0.9797373 |
3452 |
+ |
4499+ |
1.4232888 |
1.736353 |
0.3289756 |
0.8528307 |
4499 |
+ |
4512+ |
0.7377628 |
3.143991 |
0.5398056 |
0.9480726 |
4512 |
+ |
11902+ |
0.0581356 |
3.917431 |
0.9581940 |
0.9984513 |
11902 |
+ |
12203+ |
0.7146266 |
2.468814 |
0.5326933 |
0.9457086 |
12203 |
+ |
13365+ |
0.5319706 |
2.204602 |
0.6509544 |
0.9710619 |
13365 |
+ |
13441+ |
1.4779476 |
1.752619 |
0.3635666 |
0.8768117 |
13441 |
+ |
13446+ |
-0.0561098 |
2.913511 |
0.9706936 |
0.9984513 |
13446 |
+ |
13451+ |
1.0310394 |
1.583629 |
0.5285491 |
0.9456388 |
13451 |
+ |
13475+ |
-1.3589715 |
3.505009 |
0.2557154 |
0.7890100 |
13475 |
+ |
13527+ |
-0.5195980 |
2.826576 |
0.7595278 |
0.9799139 |
13527 |
+ |
13552+ |
1.4159979 |
2.451620 |
0.3564504 |
0.8744636 |
13552 |
+ |
13559+ |
0.0373990 |
2.436253 |
0.9827840 |
0.9990971 |
13559 |
+ |
Visualization
res_WT %>%
ggplot(aes(x = logFC, y = -log10(fdr), color = logCPM)) +
geom_point(alpha = 0.5) +
theme_bw() +
geom_vline(xintercept = -1, col = "red") +
geom_hline(yintercept = -log10(0.05), col = "red")
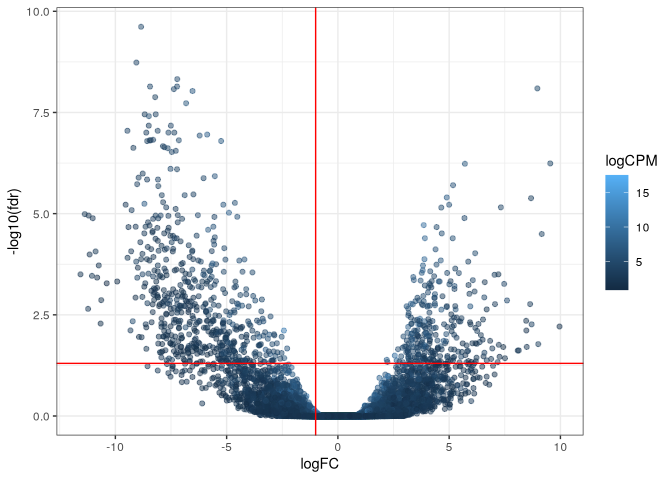
Merge comparisons and select significant positions
To identify a position as a RNase Y endonucleolytic cleavage site, Broglia et al. keep only positions where the fdr is lower than 0.01 and logFC lower of equal to -1 in both comparison we’ve made.
So we are going to merge our both result tables then filter following these criterias.
res <- res_suppl %>%
inner_join(res_WT, by = c("Position","strand"), suffix = c(".suppl", ".WT"))
Number of cleavage sites identified:
endoY <- res %>%
filter(fdr.WT<0.05 & fdr.suppl<0.05 & logFC.WT<= -1 & logFC.suppl<= -1)
endoY %>%
nrow
#> [1] 532
Number of clusters of cleavage sites identified:
endoY <- endoY %>%
arrange(Position) %>%
mutate(ID = cumsum(c(1, diff(Position) >= 5))) %>%
arrange(desc(ID))
endoY %>%
group_by(ID) %>%
n_groups
#> [1] 381
Comparison with Broglia results
We provide a small table of RNase Y cleavage sites identifed by Broglia et al. using 5’-end RNA-Seq in the extdata directory our GitLab project in order to make the comparison.
Number of RNase Y cleavage sites identified using 5’-end RNA-Seq:
csy <- read_csv("../extdata/Spyo_Broglia/RNaseY_cleavage_site_5Prime.csv", show_col_types = F)
csy %>%
nrow
#> [1] 190
Number of RNase Y cleavage sites found in common:
common <- endoY %>%
inner_join(csy, by="Position")
common %>%
nrow
#> [1] 119
Number of RNase Y cleavage sites not found by us:
not_found <- csy %>%
anti_join(endoY, by ="Position")
not_found %>%
nrow
#> [1] 71
Number of sites found not significant:
res %>%
filter(Position %in% not_found$Position) %>%
nrow
#> [1] 32
Number of sites not tested due to CPM filtering:
not_found %>%
anti_join(res, by = "Position") %>%
nrow
#> [1] 39
Notebook rendering time and memory:
time_end <- now()
rendering_time <- time_end - time_start
rendering_time
#> Time difference of 1.805431 hours
pryr::mem_used()
#> 1.61 GB