Pre-processing functions (parse_reads or demultiplex) of 5’/3’-ends RNA-Seq data
Complete source of this page is available in the notebook preprocessing.Rmd.
library(tidyverse)
time_start <- now()
library(rnaends)
The 3 datasets required for this notebook are provided in the extdata directory of the GitLab project.
Extraction of the mappable part of reads
To have a look on raw reads we want to parse, we first need to load the ShortRead package:
library(ShortRead)
Then, we inspect the reads:
fq_streamer <- FastqStreamer("../extdata/pre-processing_examples/Example_1.fastq.gz")
sr <- yield(fq_streamer)
close(fq_streamer)
sr@sread
#> DNAStringSet object of length 10000:
#> width seq
#> [1] 33 AGGAGAAGAGCGGTTCAGCAGGAATGCCGAGAC
#> [2] 33 AGGAAGAGCGGTTCAGCAGGAATGCCGAGACCG
#> [3] 33 AGGCCAGCGACGCGAAGTAGAATCAGTAATTTG
#> [4] 33 AGGCAAGGAACGCCATGCGAGAGCGGTATTATC
#> [5] 33 AGGGGTTAAGTTATTAAGGGCGCACGGTGGATG
#> ... ... ...
#> [9996] 33 AGGGCAAAAACGCGCACAAAAAATGACCAAGAA
#> [9997] 33 AGGAGGGACAGCACCGCTCTTCCGATCTTAAGC
#> [9998] 33 AGGGTCCCCCGCTTATTGATATGCAAGATGAAG
#> [9999] 33 AGGCGAGGCACGCTCAGTCAAGCTGATTTAAAT
#> [10000] 33 AGGGGGCGCGCGCTAAAAGCTACGCACGTTTTT
For this example we have 10k reads that are 33 nucleotides long that should be built that way:
1 3 4 10 11 13 14 33
AGG - VVVVVVV - CGC - XXXXXXXXXXXXXXXXXXXXX
recognition.seq - UMI - control.seq - RNA 5'-end (readseq)
The 3 first nucleotides correspond to a recognition.sequence which should always be
AGGThe 7 next nucleotides are a random sequence constituting a Unique Molecule Identifier (UMI) where the only constraint is that it must not contain any T
Then it should be an exact sequence again (control.seq) that corresponds to
CGCFinally, we have the 5’-end of the transcript
In order to extract only the part that corresponds to the 5’-end of the transcript, we have to build a read_features table using the init_read_features function:
rf <- init_read_features(start = 14, width = 19)
By default, only A, C, T or G are allowed in the nucleotidic sequence. It is possible to change the number of mismatch allowed as well as the allowed nucleotides/characters of the readseq sequence by modifying some parameters as follows:
init_read_features(start = 14, width = 19, pattern = "ACG", max_mismatch = 1)
#> # A tibble: 1 × 7
#> # Rowwise:
#> name start width pattern_type pattern max_mismatch readid_prepend
#> <chr> <dbl> <dbl> <chr> <chr> <dbl> <lgl>
#> 1 readseq 14 19 nucleotides ACG 1 FALSE
parse_reads(fastq_file = "../extdata/pre-processing_examples/Example_1.fastq.gz",
features = rf,
force = TRUE)
#> # A tibble: 2 × 2
#> is_valid_readseq count
#> <lgl> <int>
#> 1 FALSE 17
#> 2 TRUE 9983
We observe that 17 reads do not pass the ACGT (default) check for the allowed nucleotides in the readseq pattern.
fq_streamer = FastqStreamer("../extdata/pre-processing_examples/Example_1_demux/Example_1_valid.fastq.gz")
sr <- yield(fq_streamer)
close(fq_streamer)
sr@sread
#> DNAStringSet object of length 9983:
#> width seq
#> [1] 19 TTCAGCAGGAATGCCGAGA
#> [2] 19 CAGCAGGAATGCCGAGACC
#> [3] 19 GAAGTAGAATCAGTAATTT
#> [4] 19 CATGCGAGAGCGGTATTAT
#> [5] 19 TTAAGGGCGCACGGTGGAT
#> ... ... ...
#> [9979] 19 GCACAAAAAATGACCAAGA
#> [9980] 19 CCGCTCTTCCGATCTTAAG
#> [9981] 19 TATTGATATGCAAGATGAA
#> [9982] 19 TCAGTCAAGCTGATTTAAA
#> [9983] 19 TAAAAGCTACGCACGTTTT
We see that only the sequences from position 14 to 33 are present in the FASTQ output file.
Extraction of the mappable part of valid reads
In addition to what we have done before, we can extract the 5’-ends of the transcripts only for the reads which are valid by testing the validity of each expected elements described earlier.
To do that, we add some features to the read_features table with the add_read_feature function:
rf <- add_read_feature(rf,
name = "recognition.seq",
start = 1,
width = 3,
pattern = "AGG",
pattern_type = "constant")
rf <- add_read_feature(rf,
name = "control.seq",
start = 11,
width = 3,
pattern = "CGC",
pattern_type = "constant")
rf %>%
print
#> # A tibble: 3 × 7
#> # Rowwise:
#> name start width pattern_type pattern max_mismatch readid_prepend
#> <chr> <dbl> <dbl> <chr> <chr> <dbl> <lgl>
#> 1 readseq 14 19 nucleotides ACTG 0 FALSE
#> 2 recognition.seq 1 3 constant AGG 0 FALSE
#> 3 control.seq 11 3 constant CGC 0 FALSE
Following the read structure described, we define the start, the width and the expected sequence of the 2 features recognition.seq and control.seq.
As these features should have an exact sequence at the given positions, we set the pattern_type parameters = constant
In fact the pattern_type parameters can have 3 values:
nucleotidesif the pattern is a string of allowed charactersconstantif the pattern is an exact sequence (or a vector of exact sequence)regexpif the pattern is a regular expression which, once spotted, is trimmed with whatever is behind it.
Then we will add the last feature we want to check which is the UMI:
rf <- add_read_feature(rf,
name = "UMI",
start = 4,
width = 7,
pattern = "ACG",
pattern_type = "nucleotides",
readid_prepend = TRUE)
rf %>%
print
#> # A tibble: 4 × 7
#> # Rowwise:
#> name start width pattern_type pattern max_mismatch readid_prepend
#> <chr> <dbl> <dbl> <chr> <chr> <dbl> <lgl>
#> 1 readseq 14 19 nucleotides ACTG 0 FALSE
#> 2 recognition.seq 1 3 constant AGG 0 FALSE
#> 3 control.seq 11 3 constant CGC 0 FALSE
#> 4 UMI 4 7 nucleotides ACG 0 TRUE
As for other features we set the name, start and width of the feature.
As this feature is a random sequence with a list of allowed characters (A, C or G but not T) we set the pattern_type to nucleotides and the pattern to “ACG”.
We also set readid_prepend to TRUE in order to put the identified UMI in the read identifier (we will check this later).
Now that we have defined all the features, we expect to find along the reads, we can call (as in the previous example) the parse_reads function to extract the readseq subsequence of reads that have all the features valid.
report <- parse_reads(fastq_file = "../extdata/pre-processing_examples/Example_1.fastq.gz",
features = rf,
force = TRUE)
report
#> # A tibble: 10 × 5
#> is_valid_readseq is_valid_recognition.seq is_valid_control.seq is_valid_UMI count
#> <lgl> <lgl> <lgl> <lgl> <int>
#> 1 FALSE TRUE FALSE FALSE 4
#> 2 FALSE TRUE FALSE TRUE 10
#> 3 FALSE TRUE TRUE TRUE 3
#> 4 TRUE FALSE FALSE FALSE 7
#> 5 TRUE FALSE FALSE TRUE 45
#> 6 TRUE FALSE TRUE TRUE 1
#> 7 TRUE TRUE FALSE FALSE 725
#> 8 TRUE TRUE FALSE TRUE 4296
#> 9 TRUE TRUE TRUE FALSE 89
#> 10 TRUE TRUE TRUE TRUE 4820
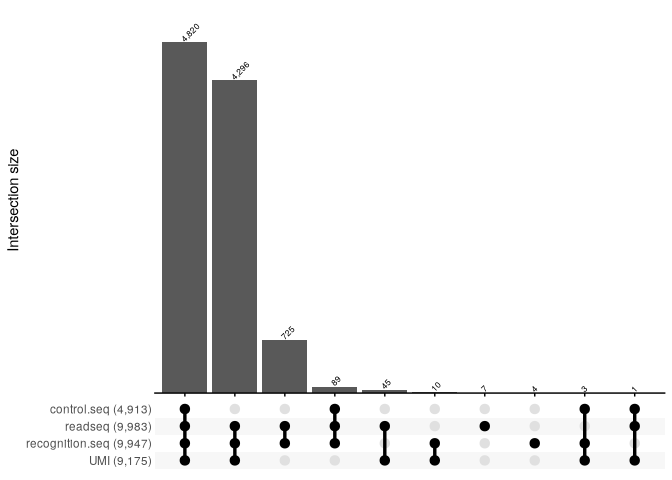
The parse_reads function returns a parse report that provides some statistics about the validity of the features checked.
For example here, we see that among the 10k input reads, 4820 were fully valid while 5163 were invalid due to an invalid feature.
We also see that most of the invalid reads have a valid recognition.seq and a valid UMI but very few reads have a valid control.seq indicating that the main reason for the invalid status of reads is due to the invalidity of the control.seq.
We can summarise these with an upset plot:
report %>%
reads_parse_report_ggupset()
In the generated FASTQ, if we see that reads only have the part of the read that corresponds to the readseq sequence:
fq_streamer <- FastqStreamer("../extdata/pre-processing_examples/Example_1_demux/Example_1_valid.fastq.gz")
sr <- yield(fq_streamer)
close(fq_streamer)
sr@sread
#> DNAStringSet object of length 4820:
#> width seq
#> [1] 19 GAAGTAGAATCAGTAATTT
#> [2] 19 CATGCGAGAGCGGTATTAT
#> [3] 19 TTCTTCCTACTAAGAACAC
#> [4] 19 TCCTCCGCTTATTGATATG
#> [5] 19 AAACGTTCAGCATTAAGTA
#> ... ... ...
#> [4816] 19 ATGTAAGTTATTTAAATAA
#> [4817] 19 CAAAACGTTAAGCGAATAA
#> [4818] 19 GCACAAAAAATGACCAAGA
#> [4819] 19 TCAGTCAAGCTGATTTAAA
#> [4820] 19 TAAAAGCTACGCACGTTTT
In the generated FASTQ, the UMI sequence was added at the beginning of each read id for later use to remove PCR duplicates:
sr@id
#> BStringSet object of length 4820:
#> width seq
#> [1] 73 CCAGCGA:SRR2017586.3.1 HWI-ST865:389:HAUHUADXX:1:1101:1718:2167 length=50
#> [2] 73 CAAGGAA:SRR2017586.4.1 HWI-ST865:389:HAUHUADXX:1:1101:1598:2170 length=50
#> [3] 73 AGCGAGA:SRR2017586.7.1 HWI-ST865:389:HAUHUADXX:1:1101:1926:2194 length=50
#> [4] 73 AGGCGAA:SRR2017586.8.1 HWI-ST865:389:HAUHUADXX:1:1101:2348:2120 length=50
#> [5] 73 ACAGGAG:SRR2017586.9.1 HWI-ST865:389:HAUHUADXX:1:1101:2374:2223 length=50
#> ... ... ...
#> [4816] 77 AACAGCA:SRR2017586.9991.1 HWI-ST865:389:HAUHUADXX:1:1101:4623:12299 length=50
#> [4817] 77 GAAGAAA:SRR2017586.9992.1 HWI-ST865:389:HAUHUADXX:1:1101:4633:12362 length=50
#> [4818] 77 GCAAAAA:SRR2017586.9996.1 HWI-ST865:389:HAUHUADXX:1:1101:4779:12322 length=50
#> [4819] 77 CGAGGCA:SRR2017586.9999.1 HWI-ST865:389:HAUHUADXX:1:1101:5003:12298 length=50
#> [4820] 78 GGGCGCG:SRR2017586.10000.1 HWI-ST865:389:HAUHUADXX:1:1101:5227:12298 length=50
Extraction of the mappable part of reads according to a barcode sequence
In order to multiplex multiple experimental conditions into one sequencing run, it is possible to ligate a barcode sequence to the ends of the transcript.
For this example we have 10000 reads that are 24 nucleotides long that should be built that way:
1 4 5 24
CAAG/TCGG - XXXXXXXXXXXXXXXXXXXX
barcode - RNA 5'-end (readseq)
The 4 first nucleotides correspond to a barcode sequence which should always be either “CAAG” or “TCGG”
Then we have the 5’-end of the transcript
fq_streamer <- FastqStreamer("../extdata/pre-processing_examples/Example_2.fastq.gz")
sr <- yield(fq_streamer)
close(fq_streamer)
sr@sread
#> DNAStringSet object of length 10000:
#> width seq
#> [1] 24 CAAGTTCAGCAGGAATGCCGAGAC
#> [2] 24 CAAGCAGCAGGAATGCCGAGACCG
#> [3] 24 CAAGGAAGTAGAATCAGTAATTTG
#> [4] 24 CAAGCATGCGAGAGCGGTATTATC
#> [5] 24 CAAGTTAAGGGCGCACGGTGGATG
#> ... ... ...
#> [9996] 24 TCGGTTAAGTTATTAAGGGCGCAC
#> [9997] 24 TCGGTAGGATGTTGGCTTAGAAGC
#> [9998] 24 TCGGTTAAGTTATTAAGGGCGCAC
#> [9999] 24 TCGGCAGCAGGAATGCCGAGACCG
#> [10000] 24 TCGGTTCAGCAGGAATGCCGAGAC
Now, we build the read_features table in order to extract the mappable part of reads and regroup them into separate output FASTQ files according to their barcode sequence.
rf <- init_read_features(start = 5, width = 20)
rf <- add_read_feature(rf,
name = "barcode",
start = 1,
width = 4,
pattern = c("TCGG","CAAG"),
pattern_type = "constant",
readid_prepend = FALSE)
rf %>%
print
#> # A tibble: 2 × 7
#> # Rowwise:
#> name start width pattern_type pattern max_mismatch readid_prepend
#> <chr> <dbl> <dbl> <chr> <chr> <dbl> <lgl>
#> 1 readseq 5 20 nucleotides ACTG 0 FALSE
#> 2 barcode 1 4 constant TCGG, CAAG 0 FALSE
Note that it is mandatory to name the feature you want to use for demultiplexing: barcode
-the start position and the width are set according to the read structure we defined,
the pattern parameter is a vector containing the 2 allowed barcode sequences “TCGG” and “CAAG”,
The pattern_type is set to “constant” because the pattern correspond to exact sequences.
Now that we have the read_feature table, we can extract the mappable part of reads that have a valid barcode sequence and split them into different output FASTQ files according to the feature named barcode using the EMOTE_demultiplex function:
report <- demultiplex(
fastq_file = "../extdata/pre-processing_examples/Example_2.fastq.gz",
features = rf,
force = TRUE
)
report
#> # A tibble: 5 × 5
#> barcode_group is_valid_readseq is_valid_barcode count demux_filename
#> <chr> <lgl> <lgl> <int> <chr>
#> 1 CAAG FALSE TRUE 17 <NA>
#> 2 CAAG TRUE TRUE 4960 ../extdata/pre-processing_examples/Example_2_demux/Example_2_CAAG_valid.fastq.gz
#> 3 INVALID TRUE FALSE 27 <NA>
#> 4 TCGG FALSE TRUE 12 <NA>
#> 5 TCGG TRUE TRUE 4984 ../extdata/pre-processing_examples/Example_2_demux/Example_2_TCGG_valid.fastq.gz
demultiplex also returns a report with statistics about the validity of features. These statistics are grouped by barcode.
Once again, if we take a look at one of the two output files, we see that we only kept the part we wanted to extract:
fq_streamer = FastqStreamer("../extdata/pre-processing_examples/Example_2_demux/Example_2_TCGG_valid.fastq.gz")
sr <- yield(fq_streamer)
close(fq_streamer)
sr@sread
#> DNAStringSet object of length 4984:
#> width seq
#> [1] 20 AGAAAAGCCAGATTTAATTA
#> [2] 20 TTTAAAGAATTAGATCAAAA
#> [3] 20 TGAATGACAATATGTCAACG
#> [4] 20 TTAAGTTATTAAGGGCGCAC
#> [5] 20 TAAGTTATTAAGGGCGCACG
#> ... ... ...
#> [4980] 20 TTAAGTTATTAAGGGCGCAC
#> [4981] 20 TAGGATGTTGGCTTAGAAGC
#> [4982] 20 TTAAGTTATTAAGGGCGCAC
#> [4983] 20 CAGCAGGAATGCCGAGACCG
#> [4984] 20 TTCAGCAGGAATGCCGAGAC
Removal of a pattern from the mappable part of reads
For this example, we have 10000 reads that have no specific structure but due to a step during the experimental protocol, reads can have a poly A sequence that should be removed in order to map on the reference genome.
fq_streamer = FastqStreamer("../extdata/pre-processing_examples/Example_3.fastq.gz")
sr <- yield(fq_streamer)
close(fq_streamer)
sr@sread
#> DNAStringSet object of length 10000:
#> width seq
#> [1] 50 TCAGCCAGTGCACGCAAAAAAAAAAAAAAAAAAAAAAAAAAAATCGGAAG
#> [2] 50 TGGGGGTAGCGAGCTTGTGGGAAGCGGTCTTTGGCGATGTGGGGGTTAAA
#> [3] 50 TTCGAATCCTACATCTGGAGCCAAAAAAAAAAAAAAAAAAAAAAAAAAAA
#> [4] 50 ATTATCCAGTCCATGTCTTTGATTATTGCCATGATGTATATTGGGGCTAA
#> [5] 50 AATTCTTGGTGTAGGGGTAAAATCCGTAGAGATCAAGAGGAAAAAAAAAA
#> ... ... ...
#> [9996] 50 CACCGCCCGTCACACCATGGGAGTTGTGTTTGCCTTAAGTCAGGATGCTA
#> [9997] 50 CATTAAAGGCGAAGGAGTCTTATACAAAAAAAAAAAAAAAAAAAAAAAAA
#> [9998] 50 ATTGGAGTGAAGGCAAATCCACCTCTGTATTTGAAAAAAAAAAAAAAAAA
#> [9999] 50 AGCACCGCTATAGGAACTCAACCTATGGTTCACCTTTGCATCAGCATTGA
#> [10000] 50 CACACCATGGGAGTTGTGTTTGCCCCAAAAAAAAAAAAAAAAAAAAAAAA
We see that several reads have a succession of As that should not map on the genome.
We will thus parse them in order to remove these poly A sequences using parse_read. For this, we first have to build a read_features table:
rf <- init_read_features(start = 1, width = 50)
For this case, the positions of the sequence to extract is variable since the locations of poly A are also variable.
We set the start position to 1 and the width to 50 (the entire read sequence), and the extraction of the mappable part will be done after the removal of the poly A. The pattern is specified as follows:
rf <- add_read_feature(rf,
name = "polyA",
start = 1,
width = 50,
pattern = "AAAAAA.*",
pattern_type = "regexp",
readid_prepend = FALSE)
rf %>%
print
#> # A tibble: 2 × 7
#> # Rowwise:
#> name start width pattern_type pattern max_mismatch readid_prepend
#> <chr> <dbl> <dbl> <chr> <chr> <dbl> <lgl>
#> 1 readseq 1 50 nucleotides ACTG 0 FALSE
#> 2 polyA 1 50 regexp AAAAAA.* 0 FALSE
As the poly A sequence can be found anywhere on the reads we set the start position to 1 and the width to 50.
The pattern corresponds to a regular expression: here, we specify a regular expression of minimum 6 A followed anything.
The pattern being a regular expression to identity and remove with whatever is behind it, we set the pattern_type to “regexp”.
With this read_features table we can perform an parse_read:
report = parse_reads(
fastq_file = "../extdata/pre-processing_examples/Example_3.fastq.gz",
features = rf,
force = TRUE)
report
#> # A tibble: 2 × 2
#> is_valid_readseq count
#> <lgl> <int>
#> 1 FALSE 1788
#> 2 TRUE 8212
The report indicates that among the 10000 reads, only 8212 were valid due to the validity of readseq.
Note that the invalidity of readseq may also be due to too short read after the removal of the poly A.
fq_streamer = FastqStreamer("../extdata/pre-processing_examples/Example_3_demux/Example_3_valid.fastq.gz")
sr <- yield(fq_streamer)
close(fq_streamer)
sr@sread
#> DNAStringSet object of length 8212:
#> width seq
#> [1] 50 TGGGGGTAGCGAGCTTGTGGGAAGCGGTCTTTGGCGATGTGGGGGTTAAA
#> [2] 22 TTCGAATCCTACATCTGGAGCC
#> [3] 50 ATTATCCAGTCCATGTCTTTGATTATTGCCATGATGTATATTGGGGCTAA
#> [4] 40 AATTCTTGGTGTAGGGGTAAAATCCGTAGAGATCAAGAGG
#> [5] 50 AATGGGGTGCACAAAGAGAAGCAATACTGCGAAGTGGAGCCAATCTTCAA
#> ... ... ...
#> [8208] 50 CACCGCCCGTCACACCATGGGAGTTGTGTTTGCCTTAAGTCAGGATGCTA
#> [8209] 25 CATTAAAGGCGAAGGAGTCTTATAC
#> [8210] 33 ATTGGAGTGAAGGCAAATCCACCTCTGTATTTG
#> [8211] 50 AGCACCGCTATAGGAACTCAACCTATGGTTCACCTTTGCATCAGCATTGA
#> [8212] 26 CACACCATGGGAGTTGTGTTTGCCCC
As we can see, poly A are removed from the reads, generating reads of different lengths.
It is possible to set a minimal length allowed in the parse_reads function (default is 18).
Notebook rendering time and memory:
time_end <- now()
rendering_time <- time_end - time_start
rendering_time
#> Time difference of 1.811553 secs
pryr::mem_used()
#> 1.57 GB